داء ولسون
| Wilson's disease | |
|---|---|
| الأسماء الأخرى | Wilson disease, hepatolenticular degeneration |
 | |
| A brown ring on the edge of the iris (Kayser–Fleischer ring) is common in Wilson's disease, especially when neurological symptoms are present. | |
| التخصص | Gastroenterology |
| الأعراض | Swelling of the legs, yellowish skin, personality changes[1] |
| البداية المعتادة | Age 5 to 35[1] |
| المسببات | Genetic |
| التشخيص المفاضل | Chronic liver disease, Parkinson's disease, multiple sclerosis, others[2][3] |
| العلاج | Dietary changes, chelating agents, zinc supplements, liver transplant[1] |
| التردد | ~1 per 30,000[1] |
داء ويلسون هو مرض وراثي يتراكم فيه النحاس الزائد في الجسم. ترتبط الأعراض عادةً بالدماغ والكبد . تشمل الأعراض المرتبطة بالكبد القيء والضعف وتراكم السوائل في البطن وتورم الساقين واصفرار الجلد والحكة . تشمل الأعراض المرتبطة بالدماغ الرعاش ، وتيبس العضلات ، وصعوبة التحدث ، وتغيرات الشخصية ، والقلق ، والذهان.[1]
ويتسبب داء ويلسون بواسطة طفرة في جين بروتين داء ويلسون (ATP7B) . ينقل هذا البروتين النحاس الزائد إلى الصفراء ، حيث يتم إفرازه في الفضلات. الحالة وراثية متنحية ؛ لكي يصاب الشخص ، يجب أن يرث نسخة محورة من الجين من كلا الوالدين. قد يكون التشخيص صعبًا وغالبًا ما يتضمن مجموعة من اختبارات الدم واختبارات البول وخزعة الكبد . يمكن استخدام الاختبارات الجينية لفحص أفراد عائلة المصابين.[1]
يُعالج داء ويلسون عادةً بالتغييرات الغذائية والأدوية. تتضمن التغييرات الغذائية اتباع نظام غذائي منخفض النحاس وعدم استخدام أدوات الطهي النحاسية. تشمل الأدوية المستخدمة عوامل خالبية مثل الترينتين ود-بنيسيلامين ومكملات الزنك . يمكن أن تشمل مضاعفات داء ويلسون الفشل الكبدي وسرطان الكبد ومشاكل الكلى . قد تكون زراعة الكبد مفيدة لمن لا تكون العلاجات الأخرى فعالة أو في حالة حدوث فشل كبدي.[1]
يحدث داء ويلسون في حوالي 1 من كل 30000 شخص.[1] تبدأ الأعراض عادة بين سن 5 و 35 سنة.[1] تم وصفه لأول مرة في عام 1854 من قبل عالم الأمراض الألماني فريدريش تيودور فون فريريتش وسمي على اسم طبيب الأعصاب البريطاني صموئيل ويلسون.[4]
العلامات والاعراض
الاماكن الرئيسية لتراكم النحاس هي الكبد والدماغ ، وبالتالي فإن أمراض الكبد والأعراض العصبية والنفسية هي السمات الرئيسية التي تؤدي إلى التشخيص.[5] يميل الأشخاص الذين يعانون من مشاكل في الكبد إلى الحصول على الرعاية الطبية في وقت مبكر ، بشكل عام كأطفال أو مراهقين ، من أولئك الذين يعانون من أعراض عصبية ونفسية ، والذين يميلون إلى أن يكونوا في العشرينيات من العمر أو أكبر. تم التعرف على البعض فقط لأن الأقارب قد تم تشخيصهم بداء ويلسون. العديد من هؤلاء ، عند اختبارهم ، تبين أنهم عانوا من أعراض الحالة ولكن لم يتم تشخيصهم..[6]
مرض الكبد
قد يظهر مرض الكبد على أنه إرهاق أو نزيف متزايد أو ارتباك (بسبب اعتلال الدماغ الكبدي ) وفرط ضغط الدم البابي . هذا الأخير ، هو حالة يزداد فيها الضغط في الوريد البابي بشكل ملحوظ ، يؤدي إلى دوالي المريء ، والأوعية الدموية في المريء التي قد تنزف بطريقة تهدد الحياة ، فضلاً عن تضخم الطحال ( تضخم الطحال ) وتراكم سائل في تجويف البطن ( استسقاء ). عند الفحص ، يمكن ملاحظة علامات مرض الكبد المزمن مثل الورم الوعائي العنكبوتي (الأوعية الدموية الصغيرة المنتفخة ، عادة على الصدر). التهاب الكبد المزمن النشط يتسبب في تليف الكبد في معظم الأوقات مع ظهور الأعراض. في حين أن معظم المصابين بتليف الكبد لديهم مخاطر متزايدة للإصابة بسرطان الخلايا الكبدية (سرطان الكبد) ، فإن هذا الخطر منخفض جدًا نسبيًا في داء ويلسون.[5]
يتم تشخيص حوالي 5 ٪ من جميع الأشخاص فقط عندما يصابون فشل كبدي حاد خاطف ، غالبًا في سياق فقر الدم الانحلالي (فقر الدم الناجم عن تدمير خلايا الدم الحمراء). يؤدي هذا إلى حدوث خلل في إنتاج البروتين (يتم تحديده عن طريق تخثر الدم المختل) والاستقلاب بواسطة الكبد. يؤدي استقلاب للبروتين المختل إلى تراكم الفضلات مثل الأمونيا في مجرى الدم. عندما تهيج الدماغ ، يصاب الشخص باعتلال دماغي كبدي (ارتباك ، غيبوبة ، نوبات ، وأخيراً انتفاخ في الدماغ يهدد الحياة.[5]
أعراض عصبية نفسية
يعاني حوالي نصف المصابين بداء ويلسون من أعراض عصبية أو نفسية. يعاني معظمهم في البداية من تدهور معرفي خفيف وكلال ، بالإضافة إلى تغيرات في السلوك. عادة ما يتبع ذلك أعراض عصبية محددة ، غالبًا في شكل باركنسن (الصمل المتقطع ، بطء الحركة أو تباطؤ الحركات ونقص التوازن هي أكثر سمات باركنسن شيوعًا[7]مع أو بدون رعاش يدوي نموذجي ، وتعبيرات وجه مقنعة ، وتداخل في الكلام ، ترنح (نقص التنسيق) أو خلل التوتر العضلي (حركات ملتوية ومتكررة لجزء من الجسم). يبدو أن النوبات والصداع النصفي أكثر شيوعًا في مرض ويلسون.[5] يصادف العديد من الأشخاص المصابين بداء ويلسون رعاشًا مميزًا يُطلق عليه "رعاش الجناح الخافق". هذا غائب عند الراحة ولكن يمكن استثارته عن طريق ابتعاد الذراعين وثني المرفقين باتجاه خط الوسط.[8]
يمكن أن يتأثر الإدراك أيضًا في داء ويلسون. يأتي هذا في فئتين ، لا يستبعد أحدهما الآخر: اضطراب الفص الجبهي (قد يظهر على شكل اندفاعية ، وضعف في الحكم علي الاشياء، عبث ، واللامبالاة ، واختلال وظيفي تنفيذي مع سوء التخطيط واتخاذ القرار) والخرف تحت القشري (قد يظهر على شكل بطء في التفكير وفقدان الذاكرة و الخلل الوظيفي التنفيذي. ، بدون علامات فقدان القدرة على الكلام أو تعذر الأداء أو عمه ). يقترح أن هذه التدخلات المعرفية مرتبطة وترتبط ارتباطًا وثيقًا بالمظاهر النفسية للمرض[7]
قد تشمل المشاكل النفسية الناتجة عن داء ويلسون التغيرات السلوكية والاكتئاب واضطرابات القلق والذهان.[5] تظهر الأعراض النفسية بشكل شائع بالاقتران مع الأعراض العصبية ونادرًا ما تظهر بمفردها. غالبًا ما يتم تحديد هذه الأعراض بشكل سيئ ويمكن أحيانًا أن تُعزى إلى أسباب أخرى. لهذا السبب ، نادرًا ما يتم تشخيص داء ويلسون عند وجود أعراض نفسية فقط..[7]
اعضاء الانظمة الاخري
تم ربط الحالات الطبية بتراكم النحاس في داء ويلسون:
- العيون: حلقات كايزر-فلايشر (حلقات KF) ، علامة مميزة مرضية ، قد تكون مرئية في قرنية العين ، إما مباشرة أو عند فحص المصباح الشقي كرواسب من النحاس في حلقة حول القرنية. إنها ناتجة عن ترسب النحاس في غشاء دسميه . يمكن أن تكون هذه الحلقات إما بنية داكنة أو ذهبية أو خضراء -محمرة ، من 1 إلى 3 ملم في العرض ، وتظهر في حوف القرنية. لا تحدث في جميع الأشخاص المصابين بداء ويلسون. يرتبط داء ويلسون أيضًا بالساد يظهر من خلال تصبغ بني أو أخضر لكبسولة العدسة الأمامية والخلفية..[9] لا يسبب أي منهما فقدان بصر كبير.[5] تحدث حلقات KF في حوالي 66٪ من الحالات التي تم تشخيصها (غالبًا في الأشخاص الذين يعانون من أعراض عصبية بدلاً من مشاكل الكبد)..[6]
- الكلى: حماض النبيبات الكلوية (النمط 2) ، وهو اضطراب في التعامل مع البيكربونات بواسطة الأنابيب القريبة يؤدي إلى الإصابة بالكلاس الكلوي (تراكم الكالسيوم في الكلى) ، وضعف العظام (بسبب فقدان الكالسيوم والفوسفات) ، وأحيانًا بيلة حمضمينية (فقدان في الأحماض الأمينية الأساسية اللازمة لتخليق البروتين).[5]
- القلب: اعتلال عضلة القلب (ضعف عضلة القلب) هو مشكلة نادرة ولكنها معروفة في داء ويلسون. قد يؤدي إلى قصور القلب (تراكم السوائل بسبب انخفاض وظيفة المضخة) وعدم اضطراب نظم القلب (نوبات من ضربات القلب غير المنتظمة و / أو السريعة أو البطيئة بشكل غير طبيعي).[5]
- الهرمونات: قصور الغدد جارات الدرقية (فشل الغدد جارات الدرقية مما يؤدي إلى انخفاض مستويات الكالسيوم) ، والعقم ، والإجهاض المتكرر.[5]
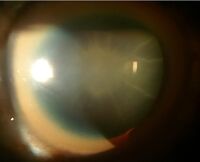
ساد العين في شكل عباد الشمس وحلقة KF السميكة لرجل يبلغ من العمر 40 عامًا مصابًا بداء ويلسون و مرض كبدي مزمن غير معوض

إضاءة منتشرة للقرنية
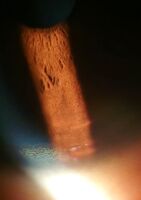
ترسب النحاس على غشاء القرنية دسميه



الجينات

يوجد جين داء ويلسون ( ATP7B ) على الكروموسوم 13 (13q14.3) ويتم التعبير عنه بشكل أساسي في الكبد والكلى والمشيمة . الرموز الجينية لإنزيم الأدينوسين ثلاثي الفوسفات من النوع P (إنزيم نقل الكاتيون) الذي ينقل النحاس إلى الصفراء ويدمجها في سيرولوبلازمين.[5] يمكن اكتشاف الطفرات في 90٪ من الحالات. معظم (60٪) متماثل الزيجوت لطفرات ATP7B (نسختان غير طبيعيتين) ، و 30٪ لديهم نسخة واحدة غير طبيعية. عشرة في المئة ليس لديهم طفرة يمكن اكتشافها.[6]
على الرغم من وصف 300 طفرة من ATP7B ، إلا أن حالات داء ويلسون في معظم السكان ترجع إلى عدد صغير من الطفرات الخاصة بهذه المجموعة السكانية. على سبيل المثال ، في المجتمعات الغربية ، توجد طفرة H1069Q (استبدال الهستيدين بالجلوتامين في الموضع 1069 في البروتين) في 37-63٪ من الحالات ، بينما في الصين هذه الطفرة غير شائعة جدًا و R778L ( أرجينين إلى ليوسين عند 778) ) في كثير من الأحيان. لا يُعرف سوى القليل نسبيًا عن التأثير النسبي للطفرات المختلفة ، على الرغم من أن طفرة H1069Q يبدو أنها تتنبأ بالظهور المتأخر والمشاكل العصبية في الغالب ، وفقًا لبعض الدراسات.[5][10] يوفر WilsonGen ، وهو مرجع شامل مشروح إكلينيكيًا ، تصنيفًا سريريًا للمتغيرات وفقًا لإرشادات ACMG و AMP الأخيرة[11]
يمكن أن يؤدي الاختلاف الطبيعي في جين PRNP إلى تعديل مسار المرض عن طريق تأخير عمر البداية والتأثير على نوع الأعراض التي تظهر. ينتج هذا الجين بروتين البريون ، الذي ينشط في الدماغ والأنسجة الأخرى ويبدو أيضًا أنه يشارك في نقل النحاس.[12] كان هناك شك في دور جين ApoE في البداية ولكن لا يمكن تأكيده.[10]
الحالة موروثة في نمط صبغي جسدي متنحي. من أجل توريثها ، يجب أن يحمل كلا الوالدين للفرد جينًا مصابًا. معظمهم ليس لديهم تاريخ عائلي لهذه الحالة..[10] يُطلق على الأشخاص الذين لديهم جين غير طبيعي واحد فقط ناقلات (متغايرة الزيجوت) وقد يعانون من تشوهات خفيفة ، ولكنها غير ذات أهمية طبية ، في استقلاب النحاس..[13]
داء ويلسون هو الأكثر شيوعًا من مجموعة الأمراض الوراثية التي تسبب فرط النحاس في الكبد. يمكن للجميع أن يسبب تليف الكبد في سن مبكر. والأعضاء الآخرون في المجموعة هم تليف الكبد الهندي في مرحلة الطفولة (ICC) ، وتليف الكبد التيرولي الطفولي المستوطن ، وتسمم النحاس مجهول السبب. لا ترتبط هذه بطفرات ATP7B: على سبيل المثال ، تم ربط ICC بالطفرات في جين KRT8 و KRT18.[10]
الفيزيولوجيا المرضية

يحتاج الجسم إلى النحاس من أجل عدد من الوظائف ، في الغالب كعامل مساعد لعدد من الإنزيمات مثل سيرولوبلازمين ، سيتوكروم سي أوكسيديز ، الدوبامين بيتا هيدروكسيلاز ، ديسموتاز فائق الاكسيد والتيروزيناز.[10]
يدخل النحاس الجسم عن طريق الجهاز الهضمي . يحمل بروتين ناقل على خلايا الأمعاء الدقيقة ، ناقل غشاء نحاسي 1 (Ctr1 ؛ SLC31A1) ، النحاس داخل الخلايا ، حيث يرتبط بعضها بالميتالوثيونين وينقل جزء منه بواسطة ATOX1 إلى عضية تعرف باسم شبكة عبر جولجي . هنا ، استجابة لتركيزات النحاس المتزايدة ، يطلق إنزيم يسمى ATP7A (بروتين مينكس) النحاس في الوريد البابي للكبد. تحمل أيضا خلايا الكبد البروتين CMT1، وميتالوثيونين وATOX1 يربط ذلك داخل الخلية، ولكن هنا ATP7B الذي يربط النحاس إلى سيرولوبلازمين ويطلقه في مجرى الدم، وكذلك إزالة النحاس الزائد عن طريق إفرازه في الصفراء . كلتا وظيفتي ATP7B معطلتان في داء ويلسون. يتراكم النحاس في أنسجة الكبد. لا يزال يُفرز السيرولوبلازمين ، ولكن في شكل يفتقر إلى النحاس (يُطلق عليه اسم أبوسيرولوبلازمين) ويتحلل بسرعة في مجرى الدم.[10]
عندما تطغى كمية النحاس في الكبد على البروتينات التي تربطه عادة ، فإنه يتسبب في أضرار مؤكسدة من خلال عملية تعرف باسم كيمياء فينتون . يؤدي هذا الضرر في النهاية إلى التهاب الكبد المزمن النشط والتليف (ترسب النسيج الضام) وتليف الكبد . يطلق الكبد أيضًا النحاس في مجرى الدم غير المرتبط بالسيرولوبلازمين. يترسب هذا النحاس الحر في جميع أنحاء الجسم ولكن بشكل خاص في الكلى والعينين والدماغ. في الدماغ ، يترسب معظم النحاس في العقد القاعدية ، خاصة في البوتامين والكرة الشاحبة (تسمى معًا النواة العدسية ) ؛ تشارك هذه المناطق عادةً في تنسيق الحركة بالإضافة إلى لعب دور مهم في العمليات الإدراكية العصبية مثل معالجة المنبهات وتنظيم الحالة المزاجية. ينتج عن الأضرار التي لحقت بهذه المناطق ، مرة أخرى بواسطة كيمياء فينتون ، الأعراض العصبية والنفسية التي شوهدت في داء ويلسون.[10]
ليس من الواضح سبب تسبب داء ويلسون في انحلال الدم ، ولكن هناك أدلة مختلفة تشير إلى أن المستوى العالي من النحاس الحر (غير المرتبط بسيرولوبلازمين ) له تأثير مباشر على أكسدة الهيموجلوبين ، وتثبيط الإنزيمات التي تزود خلايا الدم الحمراء بالطاقة. ، أو الضرر المباشر لغشاء الخلية.[14]
التشخيص
.svg&filetimestamp=20210704161647&)
قد يُشتبه في داء ويلسون على أساس أي من الأعراض المذكورة أعلاه ، أو عند اكتشاف إصابة أحد الأقارب بداء ويلسون. معظمهم لديهم اختبارات وظائف الكبد غير طبيعية مثل ارتفاع الأسبارتات ترانساميناز ، ألانين ترانساميناز ومستوى البيليروبين. إذا كان تلف الكبد كبيرًا ، فقد ينخفض الألبومين بسبب عدم قدرة خلايا الكبد التالفة على إنتاج هذا البروتين ؛ وبالمثل ، قد يطول زمن البروثرومبين (اختبار التخثر ) لأن الكبد غير قادر على إنتاج البروتينات المعروفة باسم عوامل التخثر.[5] تكون مستويات الفوسفاتاز القلوية منخفضة نسبيًا في المصابين بفشل الكبد الحاد المرتبط بويلسون.[15] إذا كانت هناك أعراض عصبية ، فعادة ما يتم إجراء التصوير بالرنين المغناطيسي (MRI) للدماغ ؛ هذا يظهر فرط كثافة في جزء من الدماغ يسمى العقد القاعدية في وضع T2..[13] قد يُظهر التصوير بالرنين المغناطيسي أيضًا سمة نمط "وجه الباندا العملاقة".[16]
لا يوجد اختبار موثوق به تمامًا لداء ويلسون ، ولكن مستويات السيرولوبلازمين والنحاس في الدم ، وكذلك كمية النحاس التي تفرز في البول خلال فترة 24 ساعة ، تُستخدم معًا لتشكيل انطباع عن كمية النحاس الموجودة في الجسم. المعيار الذهبي - أو الاختبار الأكثر مثالية - هو خزعة الكبد.[5]
سيروبلازمين

{kind=link}
{kind=link}
.svg&filetimestamp=20210704161647){kind=link}
{kind=link}
مستويات السيرولوبلازمين منخفضة بشكل غير طبيعي (<0.2g/L)في 80-95٪ من الحالات.[5] ومع ذلك ، يمكن أن يكون موجودًا بالمستويات الطبيعية لدى الأشخاص الذين يعانون من التهاب مستمر لأنه بروتين المرحلة الحادة . يوجد أيضًا انخفاض السيرولوبلازمين في مرض مينكس و فقد سيرولوبلاسمين الدم ، وهما مرتبطان بداء ويلسون ، ولكنه نادر جدًا[5][13]
يعتبر مزيج الأعراض العصبية وحلقات كايسر-فلايشر وانخفاض مستوى السيرولوبلازمين كافياً لتشخيص داء ويلسون. ومع ذلك ، في كثير من الحالات ، هناك حاجة إلى مزيد من الاختبارات..[13]
نحاس مصل الدم والبول
نحاس المصل منخفض ، وهو ما قد يبدو متناقضًا نظرًا لأن داء ويلسون هو مرض ينجم عن زيادة النحاس. ومع ذلك ، فإن 95٪ من نحاس البلازما يحمله السيرولوبلازمين ، وهو غالبًا ما يكون منخفضًا في داء ويلسون. يرتفع النحاس في البول في داء ويلسون ويتم جمعه لمدة 24 ساعة في زجاجة مع بطانة خالية من النحاس. المستويات فوق 100 ميكروغرام / 24 ساعة (1.6 ميكرومول / 24h) يؤكد داء ويلسون ، ومستويات أعلى من 40 ميكروغرام / 24 ساعة (0.6 ميكرومول / 24 ساعة) تدل بشدة..[5] إن ارتفاع مستويات النحاس في البول ليس فريدًا من نوعه في داء ويلسون ؛ يتم ملاحظتها أحيانًا في التهاب الكبد المناعي الذاتي والركود الصفراوي (أي مرض يعيق تدفق الصفراء من الكبد إلى الأمعاء الدقيقة.[13]
في الأطفال ، يمكن استخدام اختبار الپنسيلامين. 500 ملغ جرعة فموية من الپنسيلامين ، ويتم جمع البول لمدة 24 ساعة. إذا كان هذا البول يحتوي على أكثر من 1600 ميكروغرام (25 ميكرومول) ، وهو مؤشر موثوق لداء ويلسون.[مطلوب توضيح] لم يتم التحقق من صحة هذا الاختبار لدى البالغين .[13]
خزعة الكبد
بمجرد أن تشير الاستقصاءات الأخرى إلى داء ويلسون ، فإن الاختبار المثالي هو إزالة كمية صغيرة من أنسجة الكبد من خلال خزعة الكبد . يتم تقييم ذلك مجهريًا لدرجة التنكس الدهني والتليف الكبدي ، وتستخدم كيمياء الأنسجة المناعية والتقدير الكمي للنحاس لقياس شدة تراكم النحاس. مستوى 250 يميكروغرام من النحاس لكل جرام من أنسجة الكبد المجففة يؤكد داء ويلسون. من حين لآخر ، توجد مستويات أقل من النحاس ؛ في هذه الحالة ، لا يزال من الممكن أن يؤدي الجمع بين نتائج الخزعة وجميع الاختبارات الأخرى إلى تشخيص رسمي لداء ويلسون.[5]
في المراحل المبكرة من المرض ، تظهر الخزعة عادةً التنكس الدهني (ترسب المواد الدهنية) ، وزيادة الجليكوجين في النواة ، ومناطق النخر (موت الخلايا). في المرض الأكثر تقدمًا ، تكون التغييرات الملحوظة مشابهة تمامًا لتلك التي تظهر في التهاب الكبد المناعي الذاتي ، مثل التسلل عن طريق الخلايا الالتهابية ، والنخر الجزئي والتليف (النسيج الندبي). في المرض المتقدم ، أخيرًا ، يكون تليف الكبد هو النتيجة الرئيسية. في الفشل الكبدي الحاد ، يُلاحظ تنكس خلايا الكبد وانهيار بنية أنسجة الكبد ، عادةً على خلفية التغيرات التليفية. تعتبر الطرق الكيميائية النسيجية للكشف عن النحاس غير متسقة وغير موثوقة ، وتعتبر وحدها غير كافية لتحديد التشخيص.[13]
الاختبارات الجينية
يمكن إجراء تحليل الطفرات لجين ATP7B ، بالإضافة إلى الجينات الأخرى المرتبطة بتراكم النحاس في الكبد. بمجرد تأكيد حدوث طفرة ، يمكن فحص أفراد الأسرة بحثًا عن المرض كجزء من الاستشارة العائلية في علم الوراثة السريرية[5] من المهم متابعة التوزيعات الإقليمية للجينات المرتبطة بداء ويلسون ، حيث يمكن أن يساعد ذلك الأطباء في تصميم استراتيجيات الفحص المناسبة. نظرًا لاختلاف الطفرات في جين داء ويلسون بين السكان ، فإن الأبحاث والاختبارات الجينية التي يتم إجراؤها في بلدان مثل الولايات المتحدة الأمريكية أو المملكة المتحدة يمكن أن تسبب مشاكل لأنها تميل إلى أن يكون لديها مجموعات سكانية مختلطة..[17]
العلاج
النظام الغذائي
بشكل عام ، يوصى باتباع نظام غذائي منخفض في الأطعمة المحتوية على النحاس مع تجنب الفطر والمكسرات والشوكولاتة والفواكه المجففة والكبد وبذور السمسم وزيت السمسم والمحار..[5]
ادوية
تتوفر العلاجات الطبية لداء ويلسون. يزيد البعض من إزالة النحاس من الجسم ، بينما يمنع البعض الآخر امتصاص النحاس من النظام الغذائي.
بشكل عام ، يعتبر البنسيلامين هو العلاج الأول المستخدم. هذا يربط النحاس (عملية استخلاب ) ويؤدي إلى إفراز النحاس في البول. وبالتالي ، يمكن مراقبة كمية النحاس في البول لضمان أخذ جرعة عالية بما فيه الكفاية. لا يخلو الپنسيلامين من المشاكل: حوالي 20٪ يعانون من آثار جانبية أو مضاعفات لعلاج الپنسيلامين ، مثل الذئبة التي يسببها الدواء (تسبب آلام المفاصل وطفح جلدي) أو الوهن العضلي (حالة عصبية تؤدي إلى ضعف العضلات). في أولئك الذين ظهرت عليهم أعراض عصبية ، يعاني نصفهم تقريبًا من تفاقم متناقض في أعراضهم. بينما لوحظت هذه الظاهرة في علاجات ويلسون الأخرى ، فإنها عادة ما تؤخذ كمؤشر لوقف الپنسيلامين وبدء علاج الخط الثاني.[5][13] يمكن للذين لا يتحملون الپنسيلامين أن يبدأو بتراينتين هيدروكلوريد كبديلاً، والذي له أيضًا خصائص استخلابية. يوصي البعض بتراينتين كعلاج من الدرجة الأولى ، لكن التجربة مع الپنسيلامين أكثر شمولاً.[13] عامل آخر ، قيد الفحص السريري بواسطة ويلسون للادوية ، مع نشاط معروف في مرض ويلسون هو تيتراثيوموليبدات . يعتبر هذا تجريبيًا,[13] الرغم من أن بعض الدراسات أظهرت تأثيرًا مفيدًا..[5]
بمجرد عودة جميع النتائج إلى طبيعتها ، يمكن استخدام الزنك (عادة في شكل وصفة طبية من أسيتات الزنك تسمى جالازين) بدلاً من المخلبات للحفاظ على مستويات مستقرة من النحاس في الجسم. يحفز الزنك الميتالوثيونين ، وهو بروتين في خلايا الأمعاء يربط النحاس ويمنع امتصاصه ونقله إلى الكبد. يستمر العلاج بالزنك ما لم تتكرر الأعراض أو إذا زاد إفراز النحاس في البول.[13]
في حالات نادرة حيث لا تكون أي من العلاجات الفموية فعالة ، خاصة في الأمراض العصبية الشديدة ، يكون ديمركابرول (مضاد اللويزيت البريطاني) ضروريًا أحيانًا. يتم حقن هذا العلاج عن طريق الحقن العضلي (في العضل) كل بضعة أسابيع ولها آثار جانبية غير سارة مثل الألم..[18]
يتم علاج الأشخاص الذين ليس لديهم أعراض (على سبيل المثال ، أولئك الذين تم تشخيصهم من خلال فحص الأسرة أو فقط نتيجة نتائج اختبارات غير طبيعية) ، حيث قد يتسبب تراكم النحاس في حدوث أضرار طويلة المدى في المستقبل. من غير الواضح ما إذا كان هؤلاء الأشخاص يعاملون بشكل أفضل بالبنسيلامين أو أسيتات الزنك.[13]
العلاجات الفيزيائية والوظيفية
العلاج الطبيعي والعلاج الوظيفي مفيدان للمرضى الذين يعانون من الشكل العصبي للمرض. قد يستغرق العلاج بمخلب النحاس ما يصل إلى ستة أشهر لبدء العمل ، ويمكن أن تساعد هذه العلاجات في التعامل مع الرنح وخلل التوتر والرعشة ، فضلاً عن منع تطور التقلصات التي يمكن أن تنجم عن خلل التوتر العضلي
الزرع
تُعد زراعة الكبد علاجًا فعالًا لداء ويلسون ، ولكنها تُستخدم فقط في سيناريوهات معينة بسبب المخاطر والمضاعفات المرتبطة بالإجراء. يتم استخدامه بشكل رئيسي في الأشخاص الذين يعانون من فشل الكبد الخاطف والذين لا يستجيبون للعلاج الطبي أو في المصابين بأمراض الكبد المزمنة المتقدمة. يتم تجنب زراعة الكبد في حالة الأمراض العصبية والنفسية الشديدة ، والتي لم يتم إثبات فائدتها.[5][13]
المآل
إذا تُرك داء ويلسون دون علاج ، فإنه يميل إلى أن يصبح أسوأ بشكل تدريجي ويؤدي في النهاية إلى الوفاة. مع الاكتشاف المبكر والعلاج ، يمكن لمعظم المصابين أن يعيشوا حياة طبيعية نسبيًا. قد يتحسن تلف الكبد والأعصاب الذي يحدث قبل العلاج ، ولكنه غالبًا ما يكون دائمًا.[19]
التاريخ
يحمل المرض اسم الطبيب البريطاني صموئيل ألكسندر كينير ويلسون (1878-1937) ، وهو طبيب أعصاب وصف الحالة ، بما في ذلك التغيرات المرضية في الدماغ والكبد ، في عام 1912.[20] سبق عمل ويلسون واستند إلى تقارير من طبيب الأعصاب الألماني كارل ويستفال (في عام 1883) ، والذي أطلق عليه اسم "التصلب الكاذب". من قبل طبيب الأعصاب البريطاني ويليام جاورز (عام 1888));[21] قبل عالم الأمراض العصبية الفنلندي إرنست ألكسندر هومن (1889-1892) ، الذي لاحظ الطبيعة الوراثية للمرض[22] وأدولف سترومبل (1898) الذي لاحظ تليف الكبد.[21] ربط عالم الأمراض العصبية جون ناثانيال كومينجز ارتباطًا بتراكم النحاس في كل من الكبد والدماغ في عام 1948.[23] لوحظ حدوث انحلال الدم في عام 1967.[24]
في عام 1951 ، أبلغ كومينجز ، وطبيب الأعصاب النيوزيلندي ديريك ديني براون ، العاملين في الولايات المتحدة ، في وقت واحد عن أول علاج فعال ، باستخدام خالب معادن بريطاني مضاد لغاز اللويزيت.[25][26] كان لابد من حقن هذا العلاج ، لكنه كان من أوائل العلاجات المتاحة في مجال طب الأعصاب ، وهو مجال كان قادرًا بشكل كلاسيكي على المراقبة والتشخيص ولكن لم يكن لديه سوى القليل من العلاجات لتقديمه.[21][27] تم اكتشاف أول عامل إزالة معدن ثقيل عن طريق الفم ، وهو البنسيلامين ، في عام 1956 من قبل طبيب الأعصاب البريطاني جون والش.[28] في عام 1982 ، قدم والش أيضًا تراينتين,[29] وكان أول من طور رباعي الثيوموليبدات للاستخدام السريري.[30] ظهر علاج أسيتات الزنك في البداية في هولندا ، حيث استخدمه الطبيبان شوينك وهوجينراد في عام 1961 والسبعينيات على التوالي ، ولكن تم تطويره لاحقًا بواسطة بروير وزملائه في جامعة ميشيغان.[18][31]
تم توضيح الأساس الجيني لداء ويلسون ، وارتباطه بطفرات ATP7B ، من قبل العديد من المجموعات البحثية في الثمانينيات والتسعينيات.[32][33]
حيوانات اخري
تم وصف تراكم النحاس الوراثي في Bedlington Terrier,[34] حيث يؤثر بشكل عام على الكبد فقط. إنه بسبب طفرات في جين COMMD1 (أو MURR1 )..[35] على الرغم من هذه النتائج ، لا يمكن اكتشاف طفرات COMMD1 في البشر الذين يعانون من حالات تراكم النحاس غير ويلسون (مثل تليف الكبد الهندي في الطفولة ) لشرح أصلهم الجيني.[36]
انظر ايضا
مراجع
- ^ أ ب ت ث ج ح خ د ذ "Wilson Disease". NIDDK. July 2014. Archived from the original on 2016-10-04. Retrieved 2016-11-06.
- ^ Lynn, D. Joanne; Newton, Herbert B.; Rae-Grant, Alexander (2004). The 5-minute Neurology Consult (in الإنجليزية). Lippincott Williams & Wilkins. p. 442. ISBN 9780683307238. Archived from the original on 2016-11-07.
- ^ Sahani, Dushyant V.; Samir, Anthony E. (2016). Abdominal Imaging: Expert Radiology Series (in الإنجليزية) (2 ed.). Elsevier Health Sciences. p. 400. ISBN 9780323431613. Archived from the original on 2016-11-07.
- ^ "Whonamedit – dictionary of medical eponyms". www.whonamedit.com. Archived from the original on 2016-11-07. Retrieved 2016-11-06.
- ^ أ ب ت ث ج ح خ د ذ ر ز س ش ص ض ط ظ ع غ ف ق ك Ala A, Walker AP, Ashkan K, Dooley JS, Schilsky ML (2007). "Wilson's disease". Lancet. 369 (9559): 397–408. doi:10.1016/S0140-6736(07)60196-2. PMID 17276780. S2CID 24663871.
- ^ أ ب ت Merle U, Schaefer M, Ferenci P, Stremmel W (2007). "Clinical presentation, diagnosis and long‐term outcome of Wilson's disease: a cohort study". Gut. 56 (1): 115–20. doi:10.1136/gut.2005.087262. PMC 1856673. PMID 16709660.
- ^ أ ب ت Lorincz MT (2010). "Neurologic Wilson's disease" (PDF). Annals of the New York Academy of Sciences. 1184 (1): 173–87. Bibcode:2010NYASA1184..173L. doi:10.1111/j.1749-6632.2009.05109.x. hdl:2027.42/78731. PMID 20146697. S2CID 2989668.
- ^ Pagonabarraga, J; Goetz, C (2012). Biller, J (ed.). Practical Neurology (4th ed.). Philadelphia: Wolters Kluwer/Lippincott Williams & Wilkins Heath. p. 282. ISBN 978-1451142631.
- ^ Yanoff, Myron; Jay S. Duker (2008). Ophthalmology (3rd ed.). Edinburgh: Mosby. p. 411. ISBN 978-0323057516.
- ^ أ ب ت ث ج ح خ de Bie P, Muller P, Wijmenga C, Klomp LW (November 2007). "Molecular pathogenesis of Wilson and Menkes disease: correlation of mutations with molecular defects and disease phenotypes". J. Med. Genet. 44 (11): 673–88. doi:10.1136/jmg.2007.052746. PMC 2752173. PMID 17717039.
- ^ Kumar, Mukesh; Gaharwar, Utkarsh; Paul, Sangita; Poojary, Mukta; Pandhare, Kavita; Scaria, Vinod; Bk, Binukumar (2020-06-03). "WilsonGen a comprehensive clinically annotated genomic variant resource for Wilson's Disease". Scientific Reports (in الإنجليزية). 10 (1): 9037. Bibcode:2020NatSR..10.9037K. doi:10.1038/s41598-020-66099-2. ISSN 2045-2322. PMC 7270127. PMID 32493955.
- ^ Grubenbecher S, Stüve O, Hefter H, Korth C (2006). "Prion protein gene codon 129 modulates clinical course of neurological Wilson disease". NeuroReport. 17 (5): 549–52. doi:10.1097/01.wnr.0000209006.48105.90. PMID 16543824. S2CID 37186426.
- ^ أ ب ت ث ج ح خ د ذ ر ز س ش Roberts, Eve A.; Schilsky, Michael L. (2003). "A practice guideline on Wilson disease" (PDF). Hepatology. 37 (6): 1475–92. doi:10.1053/jhep.2003.50252. PMID 12774027. S2CID 263620.[dead link]
- ^ Lee GR (1999). "Chapter 48: acquired hemolytic anaemias resulting from direct effects of infectious, chemical or physical agents". In Lee GR, Foerster J, Lukens J, et al. (eds.). Wintrobe's clinical hematology. Vol. vol 1 (10th ed.). Williams & Wilkins. pp. 1298. ISBN 978-0-683-18242-2.
{{cite book}}:|volume=has extra text (help) - ^ Shaver WA, Bhatt H, Combes B (1986). "Low serum alkaline phosphatase activity in Wilson's disease". Hepatology. 6 (5): 859–63. doi:10.1002/hep.1840060509. PMID 3758940. S2CID 24055787.
- ^ Das SK, Ray K (September 2006). "Wilson's disease: an update". Nat Clin Pract Neurol. 2 (9): 482–93. doi:10.1038/ncpneuro0291. PMID 16932613. S2CID 205340375.
- ^ Ferenci, Peter (2006-06-22). "Regional distribution of mutations of the ATP7B gene in patients with Wilson disease: impact on genetic testing". Human Genetics (in الإنجليزية). 120 (2): 151–159. doi:10.1007/s00439-006-0202-5. ISSN 0340-6717. PMID 16791614. S2CID 10124665.
- ^ أ ب Walshe JM (July 1996). "Treatment of Wilson's disease: the historical background". QJM. 89 (7): 553–55. doi:10.1093/qjmed/89.7.553. PMID 8759497.
- ^ "Definition and Facts | NIDDK". National Institute of Diabetes and Digestive and Kidney Diseases (in الإنجليزية الأمريكية). Retrieved 2019-02-01.
- ^ Kinnier Wilson SA (1912). "Progressive lenticular degeneration: a familial nervous disease associated with cirrhosis of the liver" (PDF). Brain. 34 (1): 295–507. doi:10.1093/brain/34.4.295.
- ^ أ ب ت Robertson WM (February 2000). "Wilson's disease". Arch. Neurol. 57 (2): 276–77. doi:10.1001/archneur.57.2.276. PMID 10681092.
- ^ Homén EA (1892). "Eine eigenthümliche bei drei Geschwistern auftretende typische Krankheit unter der Form einer progressiven Dementia in Verbindung mit ausgedehnten Gefässveränderungen (wohl Lues hereditaria tarda)". Archiv für Psychiatrie und Nervenkrankheiten. 24: 1–38.
- ^ Cumings JN (1948). "The copper and iron content of brain and liver in the normal and in hepato-lenticular degeneration". Brain. 71 (Dec): 410–15. doi:10.1093/brain/71.4.410. PMID 18124738.
- ^ McIntyre N, Clink HM, Levi AJ, Cumings JN, Sherlock S (February 1967). "Hemolytic anemia in Wilson's disease". N. Engl. J. Med. 276 (8): 439–44. doi:10.1056/NEJM196702232760804. PMID 6018274.
- ^ Cumings JN (March 1951). "The effects of B.A.L. in hepatolenticular degeneration". Brain. 74 (1): 10–22. doi:10.1093/brain/74.1.10. PMID 14830662.
- ^ Denny-Brown D, Porter H (December 1951). "The effect of BAL (2,3-dimercaptopropanol) on hepatolenticular degeneration (Wilson's disease)". N. Engl. J. Med. 245 (24): 917–25. doi:10.1056/NEJM195112132452401. PMID 14882450.
- ^ Vilensky JA, Robertson WM, Gilman S (September 2002). "Denny-Brown, Wilson's disease, and BAL (British antilewisite [2,3-dimercaptopropanol])". Neurology. 59 (6): 914–16. doi:10.1212/wnl.59.6.914. PMID 12297577.
- ^ Walshe JM (January 1956). "Wilson's disease; new oral therapy". Lancet. 270 (6906): 25–26. doi:10.1016/S0140-6736(56)91859-1. PMID 13279157.
- ^ Walshe JM (March 1982). "Treatment of Wilson's disease with trientine (triethylene tetramine) dihydrochloride". Lancet. 1 (8273): 643–47. doi:10.1016/S0140-6736(82)92201-2. PMID 6121964. S2CID 205999334.
- ^ Harper PL, Walshe JM (December 1986). "Reversible pancytopenia secondary to treatment with tetrathiomolybdate". Br. J. Haematol. 64 (4): 851–53. doi:10.1111/j.1365-2141.1986.tb02250.x. PMID 3801328. S2CID 11546705.
- ^ Brewer GJ (January 2000). "Recognition, diagnosis, and management of Wilson's disease". Proc. Soc. Exp. Biol. Med. 223 (1): 39–46. doi:10.1046/j.1525-1373.2000.22305.x. PMID 10632959. Archived from the original on 2008-04-09. Retrieved 2008-05-20.
- ^ Bull PC, Thomas GR, Rommens JM, Forbes JR, Cox DW (1993). "The Wilson disease gene is a putative copper transporting P-type ATPase similar to the Menkes gene". Nat. Genet. 5 (4): 327–37. doi:10.1038/ng1293-327. PMID 8298639. S2CID 1236890.
- ^ Tanzi RE, Petrukhin K, Chernov I, et al. (1993). "The Wilson disease gene is a copper transporting ATPase with homology to the Menkes disease gene". Nat. Genet. 5 (4): 344–50. doi:10.1038/ng1293-344. PMID 8298641. S2CID 610188.
- ^ Sternlieb I, Twedt DC, Johnson GF, et al. (1977). "Inherited copper toxicity of the liver in Bedlington terriers". Proc. R. Soc. Med. 70 Suppl 3 (Suppl 3): 8–9. PMC 1543595. PMID 122681.
- ^ van De Sluis B, Rothuizen J, Pearson PL, van Oost BA, Wijmenga C (2002). "Identification of a new copper metabolism gene by positional cloning in a purebred dog population". Hum. Mol. Genet. 11 (2): 165–73. doi:10.1093/hmg/11.2.165. PMID 11809725.
- ^ Müller T, van de Sluis B, Zhernakova A, et al. (2003). "The canine copper toxicosis gene MURR1 does not cause non-Wilsonian hepatic copper toxicosis". J. Hepatol. 38 (2): 164–68. doi:10.1016/S0168-8278(02)00356-2. PMID 12547404.
External links
- داء ولسون at Curlie
- Wilson disease at NLM Genetics Home Reference
| Classification | |
|---|---|
| External resources |
- Articles with dead external links from February 2019
- CS1 errors: extra text: volume
- CS1 الإنجليزية الأمريكية-language sources (en-us)
- الصفحات بخصائص غير محلولة
- Short description is different from Wikidata
- Articles with hatnote templates targeting a nonexistent page
- جميع الصفحات التي تحتاج تنظيف
- مقالات بالمعرفة تحتاج توضيح from May 2016
- Articles with Curlie links
- Neurological disorders
- طب الكبد
- أمراض الكبد
- Autosomal recessive disorders
- أمراض نادرة
- النحاس في الصحة
- RTTNEURO